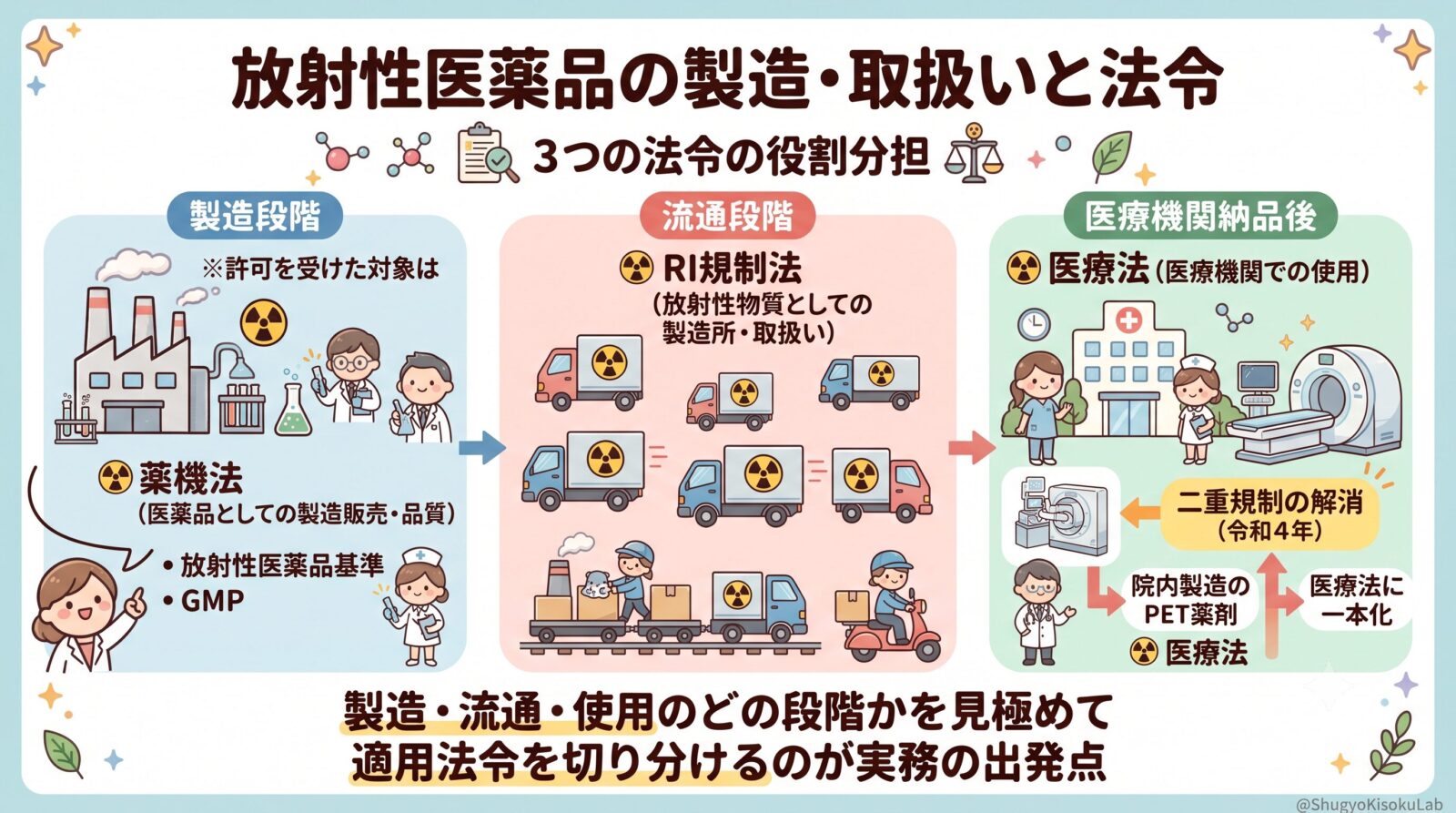
放射性医薬品は、製造から医療機関での使用まで、工程に応じて適用される法令が切り替わるという特徴があります。
薬機法・RI規制法・医療法が複雑に交錯するこの領域について、工程ごとの法令の役割分担を解説します。
放射性医薬品は「複数の法令」が工程ごとに切り替わる
放射性医薬品は、製造→流通→医療機関での使用まで、工程に応じて適用される法令が変わるのが最大の特徴です。
- 薬機法:医薬品としての製造販売・品質
- RI規制法:放射性物質としての製造所・取扱い
- 医療法:医療機関での使用(投与等)
薬機法による規制(製造段階)
放射性医薬品も「医薬品」として、製造販売の承認が必要です。「放射性医薬品基準(放薬基)」(厚労省告示)に適合することが求められ、これは製法・性状・品質・貯法等の基準です。製造所はGMP(製造管理・品質管理基準)に従います。
なお、薬機法の許可を受けた製造所に存する医薬品・原料等は、RI規制法の規制対象から除かれます。
流通から医療機関へ(販売・運搬)
承認済の放射性医薬品が製造所を出た後の「流通段階(販売・運搬)」は、RI規制法の対象となります(届出販売業の規制や、事業所外運搬の基準が適用されます)。
その後、承認済の放射性医薬品が医療機関(病院・診療所)に納品された段階で医療法の対象となり、製品としての放射性医薬品はRI規制法の適用除外となります。
医療機関での使用(医療法)
医療機関では、診療用放射性同位元素として、医療法施行規則の放射線関係規定に基づき管理されます(使用室・廃棄施設等)。また、投与された患者の退出基準(指針)にも従います。
PET薬剤(院内製造)の特殊性と二重規制の解消(令和4年)
かつては、院内でサイクロトロン等を用いて製造・調剤されるPET薬剤(未承認放射性医薬品等)は、RI規制法と医療法の二重規制の対象でした。
令和4年のRI規制法施行令改正・告示により、病院等にある未承認放射性医薬品等もRI規制法の適用除外とし、医療法に一本化されました(二重規制の解消)。これにより、医療法の枠組みで医療機関が品質を担保することになり、学会の製造施設認証等が活用されています。
なお、治験薬・グローバル治験用のものは、薬機法(治験薬GMP)下で製造されます。
むすび
放射性医薬品は、「薬機法・RI規制法・医療法」が工程ごとに役割分担する、極めて特殊な領域です。
製造段階は薬機法、流通段階はRI規制法、医療機関での使用段階は医療法というように、どの段階にあるかによって適用法令が切り替わります。
令和4年の二重規制解消も踏まえ、製造・流通・使用のどの段階かを見極めて適用法令を切り分けることが、実務の出発点となります。